BIOCHEMISTRY TOPICS
Amino acids
The general structure and properties of amino acids. Classes of amino acids. Further details on the individual amino acids.
Related: Acid-base chemistry - introduction, weak acid equilibria, polyprotic systems, titrations. Spectrophotometry.
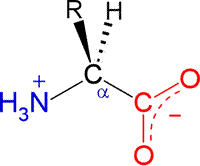
The general structural formula for an α-amino acid, shown at right, contains the amino (blue) and carboxyl (red) functional groups as substituents of a single carbon atom designated as C-alpha (Cα). These groups are shown in their expected ionization state at a physiological pH of about 7. The other two substituents of Cα are the alpha hydrogen (Hα) and a variable substituent denoted as R. When the latter is not hydrogen (nor amino or carboxyl), Cα is a chiral center. Thus the figure represents an L configuration, according to the relative configuration assignment convention. This matches the chiral configuration of the naturally-occurring α-amino acids, although exceptions are not unknown. We usually think of the natural α-amino acids as comprising a set of 20 that are encoded by nucleotide triplets and incorporated into ribosomally-synthesized polypeptides and proteins. Again, we now know of some minor, yet interesting, exceptions. The figure below shows the structural formulas for each member of the set of 20 "standard" amino acids, shown using the bond-line format. The arrangement of the structures in the figure reflects certain similarities. the two amino acids at the top left, serine (S) and threonine (T), both contain the hydroxyl functional group. Glycine (G) and proline (P) are unique in terms of chirality (Gly is achiral) and the conformational flexibility they confer upon the polypeptide chain that incorporates them. Proline could be considered to have a nonpolar character, so it is shown adjacent to the other amino acids with nonpolar hydrocarbon R groups - alanine (A), isoleucine (I), valine (V), leucine (L). Cysteine (C) and methionine (M) are the two sulfur-containing amino acids; phenylalanine (F), tyrosine (Y) and tryptophan (W) are aromatic; histidine (H), lysine (K) and arginine (R) are basic and shown in order of increasing basicity. The acidic amino acids aspartate (D) and glutamate (E), are shown together, along with their amides, asparagine (N) and glutamine (Q).
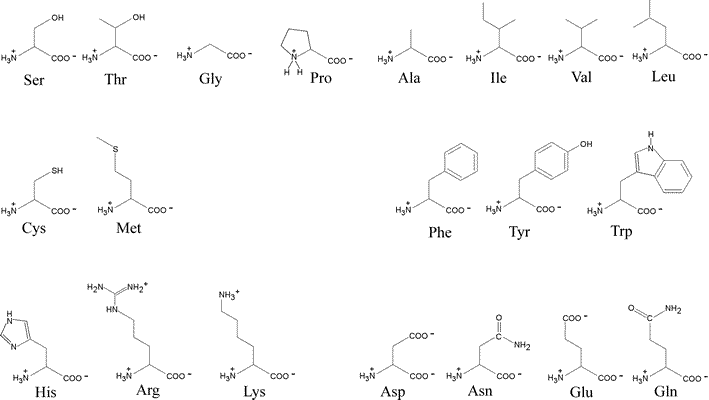
By convention, a polypeptide sequence can be represented by the single letter symbol for an amino acid, with the first letter listed the amino end or at the N-terminus and the last letter listed the carboxy end or C-terminus.
Acid-base chemistry of amino acids
The amino acids generally form diprotic or triprotic systems in aqueous solution. Amino acids that contain only the α amino and α carboxyl groups, which act as Brønsted-Lowry acid-base conjugate pairs somewhere within the normal aqueous pH range (meaning that the pKa of the acidic form of the pair lies between 0 and 14), effectively form a diprotic system, with three possible protonation or charge states. The intermediate species of a diprotic system, which can act as either an acid or a base, is in the case of diprotic amino acids, a zwitterion, or composed of two opposing formal charges. As an exercise in combining two acid dissociation equations and their associated pKa values to form a chemical equation for a proton transfer reaction and determine its Keq, we'll show that the zwitterionic form of the intermediate species is greatly favored over the form with no formal charges present. The figure below illustrates how the charge on a diprotic species (corresponding to an amino acid with an unionizable R group) varies with the "ambient" pH of the medium of which it is a component. The three possible species are positively-charged fully protonated form on the left, the intermediate form, which is a zwitterion, in the middle, and finally the fully deprotonated, negatively charged form on the right. In each of the three pH regions, 1 - 3, the species shown will be the principal (if not the only) species present. Of course, when pH = Ka1, the amounts of the fully protonated species and the intermediate, zwitterionic species will be equal. Similarly, when pH = Ka2, the amounts of the intermediate and fully deprotonated forms will be equal.
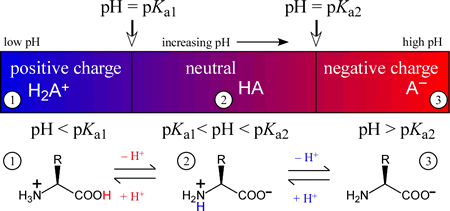
Note that one possible form of a diprotic amino acid is missing from the principal species versus pH diagram above is the neutral form with a protonated carboxyl group and a neutral amino group, i.e. H2NCHRCOOH. We can show that zwitterionic form would predominate to such a great extent over this form that it could only exist in solution in negligible amounts. Using glycine as an example, with a carboxyl group pKa of 2.34 and 9.60 as the pKa of its protonated amino group, we can calculate the equilibrium constant for the proton transfer reaction:
1 H2NCH2COOH → H+ + H2NCH2COO−
2 H+ + H2NCH2COO− → +H3NCH2COO−
sum: H2NCH2COOH → +H3NCH2COO−
The equilibrium constant for such a proton transfer reaction is calculated as follows (with the subscript 1 denoting the carboxyl group and 2 denoting the amino group):
pKeq = pKa1 − pKa2 = 2.34 − 9.60 = −7.26
Keq = 10−pKeq = 10+7.26 = 1.8 × 107
This equilibrium greatly favors the zwitterion. Hence we conclude that equilibrium amounts of a fully neutral intermediate species of a diprotic amino acid are negligible in comparison to the zwitterionic form.
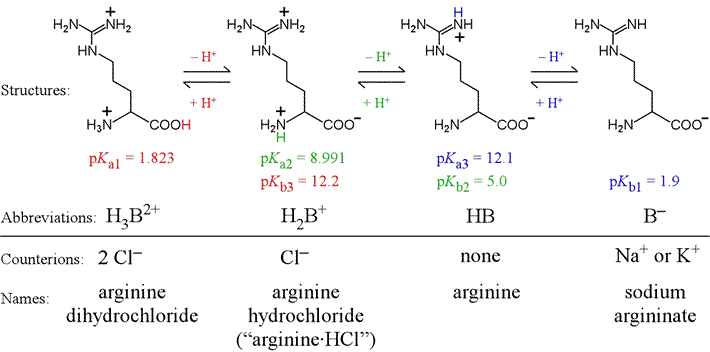
Triprotic amino acids contain an additional group as part of its variable R group that acts in acid-base equilibrium within the typical aqueous pH range. In this case, which is similar to phosphate, there are two intermediate forms. As an example of a triprotic amino acid, let us consider arginine. Arginine has the most basic R group of the 20 amino acids, due to its guanidinium group. There are three pKa values associated with the different protonation/charge states of arginine, illustrated in the figure below. Arginine is here shown in a bond-line format.
More on amino acids. From here it is customary to inquire about the biochemical and other biologically relevant properties of the individual amino acids. Since so much of our interest in them concerns their role in proteins, it is important to point out that in this respect, the amino acids exist as residues within a long linear polymer, with each amino acid linked to the main chain by peptide bonds and contributing its distinctive R group as its side chain. From the perspective of acid-base chemistry, apart from the terminal amino acids, only the R groups would contribute to the acid-base properties of a polypeptide chain. From an analytical viewpoint, it would be relevant to consider the spectroscopic properties of the peptide group and individual amino acid side chains.
Other perspectives on amino acids include their metabolic and other biochemical roles. such as neurotransmitters, hormone precursors, and their many biological modifications. There is a rich and fascinating body of inquiry into the secondary metabolism of amino acids, which gives rise to a variety of natural products.
Glycine
Glycine (Gly, G) is the simplest of the 20 naturally-occurring amino acids. As noted above, since R is just a hydrogen, glycine is the only natural amino acid that is not chiral at the alpha carbon. Although in some classification schemes, glycine is considered nonpolar, hydrogen is so small that it contributes negligibly to nonpolar surface area. It is much more significant that the smallness of its hydrogen R group offers relatively little steric hindrance to bond rotations at Cα and thus the presence of Gly confers greater conformational flexibility in the context of a polypeptide chain.
Histidine
Histidine (His, H) is one of the most interesting amino acids because of the variety of roles it can play in protein function, especially as a key residue in many enzyme active sites. Of all the ionizable side chains, the typical pKa of the imidazole ring of His is closest to a neutral pH. Studies of model compounds have established a range of 6.0 - 7.0 for the intrinsic pKa of the histidine side chain.
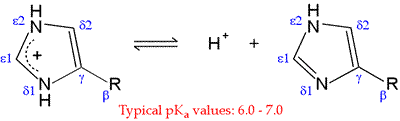
The neutral form of the imidazole ring can exist in two different tautomeric forms: with hydrogen on the δ1 nitrogen or with hydrogen on the ε2 nitrogen. The pKa of the ε2 nitrogen has been shown in 13-C NMR studies of a model compound to be about 0.6 pH units higher than that of the δ1 nitrogen, so in the absence of countervailing environmental effects, the form on the right will tend to predominate.

Neutral imidazole is a particularly good nucleophile, and histidine is one of the more reactive residues in proteins. With a pKa near 7, the imidazole side chain is one of the strongest bases that can exist at neutral pH. In its neutral form, the imidazole side chain has an "ambidextrous" nature, since the nitrogen without a hydrogen is nucleophilic and can act as a hydrogen bond acceptor, while the nitrogen with the hydrogen bond is electrophilic and can act as a H-bond donor.
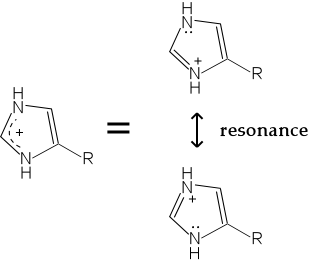
Protonation of a histidine residue inactivates it as a nucleophile. The protonated form of the imidazole ring is stabilized by resonance, by which the positive charge is shared by both nitrogen atoms of the ring.
A prominent example of histidine as a crucial catalytic component in an enzyme mechanisms found in the serine proteases. Histidine is the central residue in a catalytic triad that is characteristic of this type of enzyme. A neutral imidazole acts as a base to enhance the power of serine as a nucleophile to attack the acyl carbon of a peptide bond to form a tetrahedral intermediate. The protonated His residue in turn acts as a proton donor (general acid) to promote the loss of a leaving group from a tetrahedral intermediate.
Cysteine
Cysteine (Cys, C), one of two sulfur-containing amino acids, bears the most reactive side chain, a thiol (-SH, also called sulfhydryl) group attached to the beta carbon. The thiol is weakly acidic (intrinsic pKa 9.0-9.5), its the dissociation leaves the thiolate anion. Both of these, particularly thiolate, are good nucleophiles, so the cysteine side chain can engage in many substitution reactions. Other reactions involve the oxidation of the thiol group.
The nucleophilic thiol group can be alkylated by reaction with alkyl halides or iodoacetate. Another common reaction, especially important one for cysteine's biological role in protein function, is the formation of a thioester linkage (formally a carboxylic acid derivative).
A disulfide bond is a covalent chemical bond between two sulfur atoms that can arise from the oxidative linking of two sulfhydryl (thiol) groups. This is a common theme for cysteine residues in proteins, especially those in oxidizing environments such as prevailing extracellular conditions. The formation of disulfide bonds within proteins in vivo is a common example of a posttranslational modification.
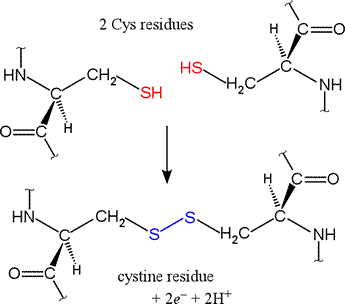 Shown at right are two cysteine residues in polypeptide chain(s).
The thiol groups are in their reduced forms (in red in figure).
Removal of two hydrogens (H+ + e−) from each thiol
(by an oxidizing agent, not included in the figure, which represents an oxidation half-reaction),
and concomitant formation of a new covalent bond - the disulfide bond -
between the two sulfur atoms yields the lower structure at right.
A disulfide-linked pair of cysteine residues is termed a cystine residue.
The conversion of two sulfhydryl groups to a disulfide
linkage is an oxidation reaction. Conversely, disulfide bonds can
be reduced to yield two thiols, which is the reverse of the half-reaction shown at right.
Reagents used to break disulfides notably include other thiol-containing species, such as
β-mercaptoethanol (mercaptan is yet another name for the thiol group) and
glutathione. The reduced thiols undergo a disulfide exchange reaction with disulfide-linked species.
Shown at right are two cysteine residues in polypeptide chain(s).
The thiol groups are in their reduced forms (in red in figure).
Removal of two hydrogens (H+ + e−) from each thiol
(by an oxidizing agent, not included in the figure, which represents an oxidation half-reaction),
and concomitant formation of a new covalent bond - the disulfide bond -
between the two sulfur atoms yields the lower structure at right.
A disulfide-linked pair of cysteine residues is termed a cystine residue.
The conversion of two sulfhydryl groups to a disulfide
linkage is an oxidation reaction. Conversely, disulfide bonds can
be reduced to yield two thiols, which is the reverse of the half-reaction shown at right.
Reagents used to break disulfides notably include other thiol-containing species, such as
β-mercaptoethanol (mercaptan is yet another name for the thiol group) and
glutathione. The reduced thiols undergo a disulfide exchange reaction with disulfide-linked species.
Perturbed or "anomalous" pKa values
As has already been suggested, the intrinsic pKa values for ionizable groups are no guarantee that a particular residue in a particular protein will be in a particular ionization state at a pH consistent with its physiologically relevant structure and function. Some pKa values are perturbed significantly from their intrinsic values, and these "anomalous" values are furthermore demonstrably important for the proper function of a protein in some cases.
To illustrate the idea, consider an aspartate residue in a neutral (pH 7) aqueous environment with a "normal" pKa. The residue will be overwhelmingly ionized. By plugging in values for the pKa of the residue, the pH of the medium, the Henderson-Hasselbalch equation can be used to calculate the proportion of ionized to unionized forms of the residue. The top half of the figure below illustrates the situation.
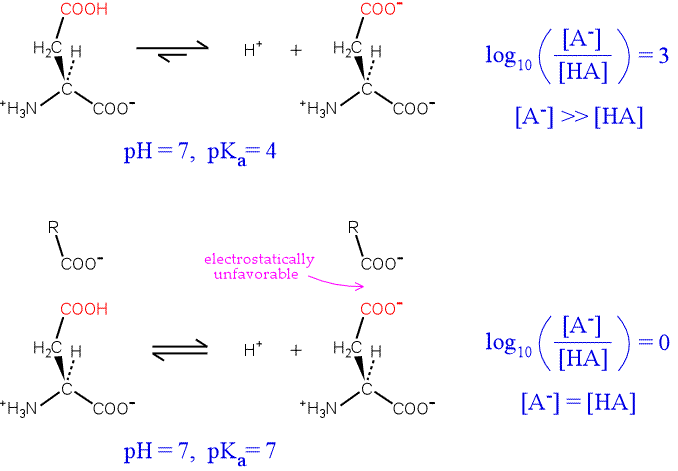
Now consider the influence of a nearby negative charge on the ionization of our hypothetical residue. The presence of the negative charge makes the ionization much less favorable, shifting the equilibrium to the left. The greater the shift in the equilibrium, the more the pKa is raised from its intrinsic value. In the extreme case illustrated, the amounts of both forms of the residue are equal, and the pKa has been perturbed upward by three units.
An example of this effect where the residue with an anomalous pKa is directly involved in the protein's function is provided by lysozyme. Lysozyme is an enzyme produced by a variety of organisms that hydrolyzes the polysaccharide component of the peptidoglycan cell walls of many types of bacteria. The mechanism of lysozyme depends on two acidic residues, Asp52 and Glu35. Asp52 has a normal pKa. Its negative charge stabilizes a developing positive charge as the reaction proceeds through a oxonium ion intermediate. However, Glu35 has an anomalously high pKa (its pKa is thought to be about 6.5), keeping it more in the protonated form. The unionized form of Glu35 donates a hydrogen ion to the oxygen of the glycosidic linkage, assisting the breaking of the bond between sugar residues. Glu35 would not be so effective in this role if its pKa was normal.
Amino acid derivatives
Tyrosine can be readily derived from phenylalanine by hydroxylation of the latter. Thus, while phenylalanine is an essential amino acid, tyrosine is not. Further hydroxylation of tyrosine, combined with its decarboxylation leads to the catecholamine neurotransmitter dopamine, which in can be further derivatized to norepinephrine and epinephrine.
Posttranslational modifications of amino acid residues
Phosphorylation and glycosylation are very common posttranslational modifications. The most commonly phosphorylated residues in proteins are serine, threonine, and tyrosine. There are very many types of posttranslational modifications. Some other notable examples are the hydroxylation of proline, which is crucial in collagen, and the carboxylation of glutamate to form γ-carboxyglutamate, particularly adept at chelating Ca2+, which is important to the cascade of protein factor activation in blood clotting.
A particularly dramatic (and colorful!) example of posttranslational modification is the green fluorescent protein (GFP), a bioluminescent jellyfish protein. GFP undergoes a spontaneous reaction that converts a three-residue sequence, Ser-Tyr-Gly, into a fluorophore.