Lecture 7. Protein structure
Thursday 7 February 2019
Introduction to the three-dimensional structure of proteins. Structure and characteristics of the peptide group. Dihedral angles (ω, φ, ψ) and Ramachandran plots. Secondary structure: α helix and β sheet. Secondary structure connections: turns, motifs. Fibrous protein structure: The α-helical coiled coil and keratin. Secondary structure propensities of amino acid residues.
Reading: Lehninger - Ch.4, pp.115-142.
Summary
Reading summary. §4.1 Overview of protein structure. A protein's structure is stabilized largely by weak interactions. The peptide bond is rigid and planar. §4.2 Protein secondary structure. The α helix is a common protein secondary structure. Table 4-1 Idealized φ and ψ angles for common secondary structures in proteins. Box 4-1 -Methods- Knowing the right hand from the left. Amino acid sequence affects stability of the α helix. Table 4-2 Propensity of amino acid residues to take up an α-helical conformation. The β conformation organizes polypeptide chains into sheets. β turns are common in proteins. Common secondary structures have characteristic dihedral angles. Common secondary structures can be assessed by circular dichroism. §4.3 Protein tertiary and quaternary structures. Fibrous proteins are adapted for a structural function. α-keratin, collagen. Box 4-2 Permanent waving is biochemical engineering. Box 4-3 -Medicine- (why everyone should eat fresh fruits and vegetables) Silk fibroin. Structural diversity reflects functional diversity in globular proteins. Box 4-4 The Protein Data Bank. Globular proteins have a variety of tertiary structures. Motifs, supersecondary structure. Box 4-6 -Methods- methods for determining the three-dimensional structure of a protein (pp.134-136). Domains. Rules derived from studies of common protein folding patterns. Some proteins or protein segments are intrinsically disordered. Protein motifs are the basis for protein structural classification. Topology diagrams. Protein quaternary structures range from simple dimers to large complexes.
***
Key terminology and resources
A polypeptide chain is formed by linking amino acids together in peptide bonds in a linear, unbranched manner. By convention, such a polypeptide chain is "read" from the amino acid with a free α-amino group (which is denoted the N-terminal residue) to the residue with a free α-carboxyl group (which is denoted the C-terminal residue). The conformation of a polypeptide chain can be specified by a series of dihedral, or torsion angles.
Structural hierarchy: The hierarchical description of protein structure begins with primary structure, which relates to the covalent description of a (possibly modified) polypeptide. Modifications aside, the primary structure of a polypeptide chain is determined by its amino acid sequence. Secondary structure is a "local" (in the sense of actual three-dimensional space) description of the conformation of a polypeptide chain. Secondary structures show regular patterns, the two prominent examples being the α helix (alpha helix) and the β sheet (beta sheet).
Tertiary structure is the overall shape of the polypeptide chain, which depends on the conformation of all the main chain and side chain bonds of the molecule. Quaternary structure refers to the cases in which separate polypeptide chains ("monomers" or "subunits") associate - usually noncovalently - to form "multimers" or "oligomers".
Exploring protein structures. A wealth of protein structure information is readily accessible through the Protein Data Bank (PDB). There are a variety of tools for visualizing and manipulating the structures found there. The PyMOL molecular graphics program is an especially useful and powerful tool, and the images of protein structure representations seen on this website were produced using it.
The peptide bond
Peptide bonds are the amide bonds that link amino acids together to form peptides (short chains of amino acids) and proteins (longer polypeptide chains that fold into specific three-dimensional conformations with biological function). It is important to understand the chemical and structural characteristics of peptide bonds, thus their outstanding features are described here.
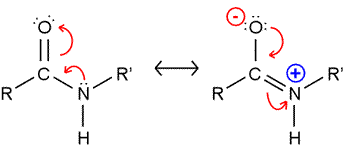
The peptide bond presents a significant barrier to rotation. The second resonance structure of the two drawn in the figure shown at left is invoked to explain this, and bond length comparisons are consistent with partial double bond character. As a consequence, the atoms shown in the figure are all constrained to lie in the same plane. (Note that in polypeptide chains, R and R' represent Cα atoms and their attached groups.)
Peptide bonds: trans and cis forms
The peptide bond as we've seen is planar. But the planar conformation can be accommodated in two alternate forms denoted as trans and cis. The cis form is less stable because of its greater steric repulsion between the Cα atoms and their attached groups. Therefore, trans peptide bonds predominate in proteins.
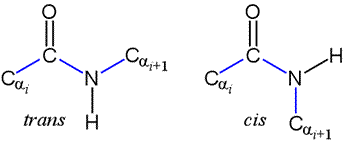
Note that a Newman projection of the C-N bond would show two dihedral angles: one in which the Cα atoms are anti (180°), corresponding to trans, and one in which the Cα atoms are eclipsed (0°), which is cis. This dihedral angle is called omega (ω).
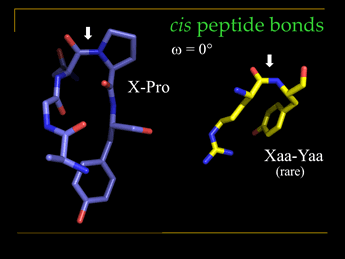
It will serve well to contrast X-Y peptide bonds with X-Pro peptide bonds. Here X and Y represent generalized amino acids. As noted, the trans configuration better avoids steric clash between Cα substituents on neighboring residues, either the side chain or the main chain. However, for the case of X-Pro, this difference is much reduced due to the unique ring structure of a Pro residue, its side chain also joined at the amide nitrogen of the peptide bond. Only about 0.03% of X-Y peptide bonds (Y is any non-Pro residue) are cis, whereas about 5% of X-Pro peptide bonds in protein structures are cis. The presence of cis peptide bonds is often associated with important functional properties.
Polypeptide chains
A polypeptide main chain, or "backbone" is made up of a repeating three-atom unit: the amide nitrogen (often denoted simply as N in this context), the alpha carbon (Cα), and acyl carbon (C′, or sometimes just C). The repeating functional groups, amide and acyl, give the main chain a polar character. The main chain amide (-NH- group) is a hydrogen bond donor, and the main chain acyl group ( >C=O group) is a hydrogen bond acceptor.
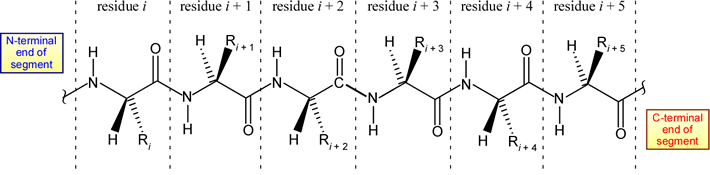
The structural representation of a six-residue polypeptide segment shown above is meant to be a stereochemically correct representation of L-amino acids linked by trans peptide bonds (which are intersected by the dashed vertical lines).
Dihedral angles (ω, φ, ψ) and Ramachandran plots
In order to describe the conformations of polypeptide chains, torsion angles (also called dihedral angles) are employed. Dihedral angles are defined by four atoms in a chain connected by three bonds with the angles about the central bond ranging over the interval [−180°, 180°]. The conformation of the polypeptide main chain can be specified by a succession of three repeating dihedral angles. The planar conformation of the peptide bond results in the restriction of its associated dihedral angle (denoted as omega, ω). The two other defined angles, phi (φ) and psi (ψ) are more freely variable since their central bonds are purely single bonds (from N to Cα and from Cα to the acyl carbon, respectively). The pairs of (φ, ψ) values for residues in a protein are commonly plotted graphically in a representation of polypeptide chain conformation called a Ramachandran plot.
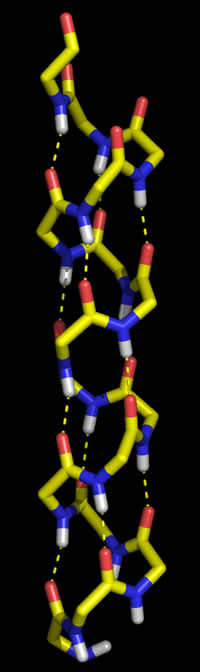
Secondary structures: the α helix and β strand conformations
Polypeptide chains can adopt regularly repeating main chain conformations. Such conformations are generally the most stable (energetically favorable). One of these conformations, proposed first by Pauling and in fact observed prominently in protein tertiary structure, is the α helix (alpha helix, model at right).
Right: Molecular graphics image of 16 residues of poly Gly in the α helix conformation. In the stick representation, atoms are at ends and vertices, and yellow indicates carbon, red oxygen, blue nitrogen, and white hydrogen (Only main chain amide hydrogen atoms are shown). Hydrogen bonds are indicated by the dashed yellow lines, and form between the acyl oxygen of residue i (acceptor) and the amide group of residue i + 4 (donor).
The other regular conformation is the β strand conformation. Together with turns and loops, the α helix and β strand could be likened to "elements" that are put together to form higher-order "compounds" of protein structure.
Characteristics of the α helix
The ideal α helix has the following features:
- Right-handed
- 3.6 residues per turn
- Pitch = 5.4 Å
- H-bond distances (amide N to acyl O) ~ 2.8 Å
- In proteins, an average length is ~ 12 residues (~ 18 Å)
β strands and sheets
Compared to the α helix, the β strand conformation yields a much more extended main chain. A fully-extended main chain would correspond to both φ and ψ dihedral angles close to ±180°. While this extension forecloses the possibility of intrachain interactions between main chain groups, it permits hydrogen bonding between these groups in juxtaposed β strands. The strands can align in either parallel or antiparallel orientation, and juxtaposition of more strands gives rise to what are termed β sheets.
The figure below illustrates the beta conformation and the scheme for hydrogen bonding between β strands. From upper left, clockwise: A three-residue extended conformation; the schemes for cross-strand H-bonds for adjacent strands in antiparallel and in parallel orientations; and finally (bottom left) two representations of a real four-stranded antiparallel β sheet (from a structure for a human antibody) (a) ribbon representation, (b) stick model. In both cases, the strands are shown in yellow, connecting loops in blue.
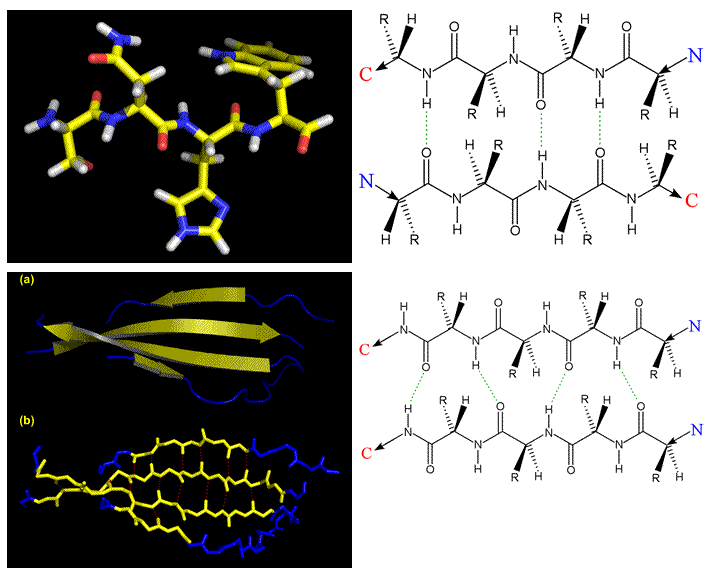
Characteristics of the β conformation
Ideal parameters for β strands are as followings:
- Antiparallel β sheet: φ = −139°, ψ = +135°, ω = −178°
- 2 residues per turn
- Pitch = 6.8 Å
- Parallel β sheet: φ = −119°, ψ = +113°, ω = 180°
- 2 residues per turn
- Pitch = 6.4 Å
Connecting 2° structure elements: Turns
The architecture of globular proteins tends to be characterized by secondary structural elements that crisscross or traverse the molecule, and connecting segments where the direction of the polypeptide chain reverses, and the next 2° structure segment leads back toward the center of the molecule. These connecting segments - short turns and longer loops - thus tend to occur at the surface of the globular protein. Often such turn or loop segments are quite important for the protein's function.
Short turns, where the direction of the main chain changes radically, can be quite varied and show unusual conformations, although some forms recur quite frequently in protein structures. The most common short turns are well exemplified in β-hairpins (see below), in which two adjacent antiparallel β strands are linked.
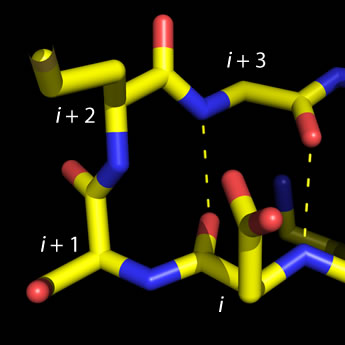 Left: Molecular graphics image of a type I beta turn.
Residue positions i through i + 3 are labeled,
and H-bonds between main chain groups of i and i + 3 are indicated by the dashed lines.
The four-residue sequence for this turn is ESMG (residue i is Glu and
residue i + 3 is Gly).
Left: Molecular graphics image of a type I beta turn.
Residue positions i through i + 3 are labeled,
and H-bonds between main chain groups of i and i + 3 are indicated by the dashed lines.
The four-residue sequence for this turn is ESMG (residue i is Glu and
residue i + 3 is Gly).
The type defined by four residues (designated i, i + 1, i + 2, and i + 3) and called β (beta) turns, or reverse turns, (also called β bends in some texts), fit the following pattern: The acyl oxygen of residue i makes a hydrogen bond with the amino group of residue i + 3, while main chain groups of the central residues (i + 1 and i + 2) are not involved in the H-bonding pattern of an antiparallel β sheet. While four subtypes of β turns have been described, for most commonly observed of them glycine predominates in residue position i + 3, and Pro in position i + 1. Furthermore, Asp, Asn, and Ser are common at position i, since their side chains can make favorable interactions (H-bonds) with main chain groups of the central residues.
In the less-common γ (gamma) turn, the acyl oxygen of residue i makes a hydrogen bond with the amino group of residue i + 2.
Simple motifs and topological representations
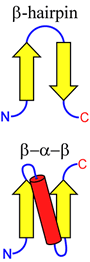
The figure at right illustrates two simple ways in which β-strand elements can be connected. This more abstract way of representing protein structural features is called a topology diagram. Note that the β-hairpin motif aligns the connected β-strands in an antiparallel orientation, whereas an α-helix (or an extended loop) can be used to connect β-strands in a parallel fashion, by forming a β-α-β motif.
These very simple motifs can be repeated and elaborated into larger motifs, or supersecondary structure, as will be illustrated further.
"Helical wheel" diagram and amphipathic helices
In terms of primary structure, β sheets can be made up of nonlocal β strand alignments. Beta strands from different polypeptide chains can be juxtaposed to form a sheet that is continuous across a subunit interface in a quaternary structure. Thus, we begin to visualize how secondary structure "elements" build up higher level structure through nonlocal interactions. Switching our focus to α helices, we can observe nonlocal associations between helices that build up higher level protein structure. In contrast to β sheets, in which β strand associations are made vis H-bonding between main chain polar groups, α helices associate due to favorable contacts between side chains.
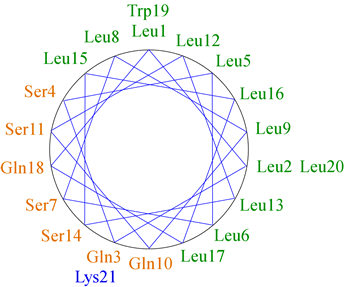
The figure at left shows the positions of the side chains along a regular α-helix. For a right-handed helix with 3.6 residues per turn, rotation is clockwise as the polypeptide chain is followed N to C, and the 5th residue ends up in a position 40° clockwise relative to residue 1. Note that over a number of turns of the helix (21 residues or nearly six turns of the helix), a pattern of distribution of the side chains emerges in this example. Nonpolar side chains group on one side of the helix, whereas polar and charged side chains are grouped to the other side. This gives the helix an amphipathic character, having obvious implications for the orientation of such a helix in the context of tertiary structure. The nonpolar face of the helix can interact favorably with (and form part of) the nonpolar core of a globular protein, while the hydrophilic face will orient toward the surface.
A slight variation on this idea leads to the coiled coil motif, which occurs in the structural protein keratin and in many other instances. In this case, associations between α helices is based largely upon nonpolar interactions made possible by a heptad repeat sequence pattern (see the α helix topics page for further details).
Residue propensities
Secondary structure propensities for amino acid residues. The following data is reproduced from Creighton (1993).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related topics pages:
- dihedral or torsion angles
- secondary protein structure
- circular dichroism (CD) spectroscopy
- tertiary protein structure